
|
||||||||
Établir une phylogénie
Établir une phylogénie c'est retrouver les liens de parenté entre différents êtres vivants. Ces liens de parentés sont par la suite représentés graphiquement dans des arbres phylogénétiques.
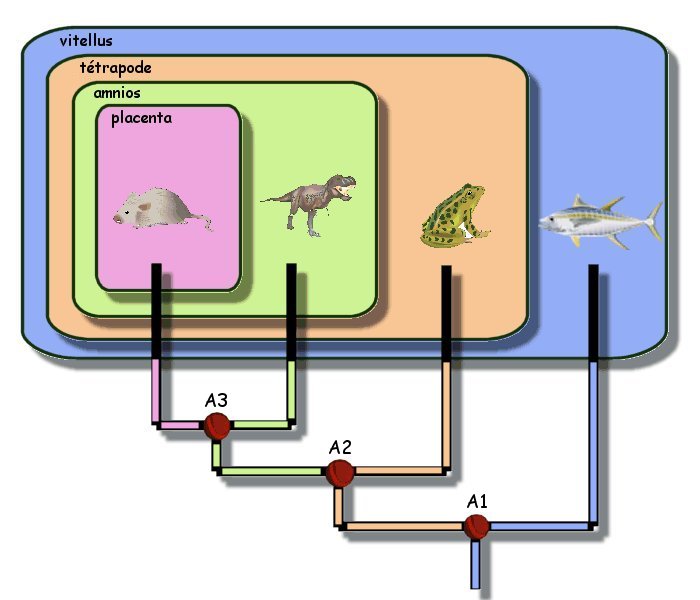
Un caractère ou un état de caractère peut être qualifié de dérivé ou bien d'ancestral. Un caractère ancestral est un caractère partagé par deux êtres vivants et donc qui existait chez leur ancêtre commun. un caractère dérivé sera, par conséquent, un caractère qui n'existe que chez l'une ou l'autre des deux espèces apparentées. La dénomination "ancestral" ou "dérivé" pour un caractère donné est donc relative et ne peut s'utiliser qu'en parlant d'espèce précises. En effet, un caractère peut être considéré comme ancestral pour certaines espèces mis dérivé pour d'autres. En reprenant l'exemple figurant dans l'image ci-dessus :
En définitive, la désignation d'un état dérivé ou ancestral pour un caractère n'est valable que pour une phylogénie donnée et construite. Il est également intéressant de comprendre que seuls les états dérivés permettent l'obtention de différents groupes et de créer des apparentements. Dans l'exemple déjà utilisé, il aurait été impossible de créer des emboîtements uniquement avec des caractères partagés par tous (les 4 animaux seraient tous dans une même boite et donc impossibles à apparenter les uns par rapport aux autres) : c'est bien la présence de caractères dérivés qui a permis de constituer des emboîtements de plus en plus restrictifs.
Il existe des molécules communes aux êtres vivants ayant des ancêtres communs. On cherche à regarder les différences dans ces molécules pour établir des phylogénies moléculaires. On parle d’ailleurs de molécules homologues s’il y a similitude des séquences nucléotidiques et/ou polypeptidiques (au moins 20% d’homologie ou de similitude des séquences). Toutes les molécules ne sont pas utilisables pour établir des liens de parenté : seules celles pouvant à la fois se " transmettre " à la descendance et " évoluer " sont utilisables. De ce fait, seul l’ADN est utilisable. On y adjoint la possibilité d’utiliser les polypeptides qui, bien que non transmis directement d’une génération à l’autre pour la plupart, sont issus de la transcription et de la traduction des gènes de l’ADN ; il sont des indices imparfaits des séquences nucléotidiques de l’ADN (" l’imperfection vient de la redondance du code génétique pour certains acides aminés). Autre limite à l’utilisation des molécules : il faut que la molécule étudiée soit partagée par un grand nombre d’individus ou d’espèces puisqu’il s’agit d’établir des liens de parenté. Plus la molécule est partagée par un grand nombre, plus les relations de parentés seront vastes.
Un exemple : établir des liens de parentés à partir de la comparaison des séquences des globines a des vertébrés. L’hémoglobine est une protéine formée de quatre sous unités appelées globines. Chaque globine correspond donc à une chaîne précise en acides aminés (qu’il est possible de séquencer) associée à un hème (partie non polypeptidique permettant le transport d’une molécule de dioxygène).
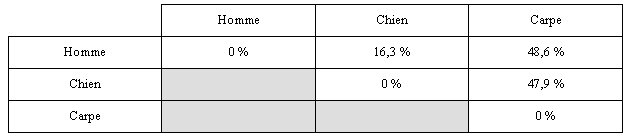
Les différences correspondent au nombre d’acides aminés différents quand on compare des séquences deux à deux. Ces valeurs peuvent être reportées dans un tableau des différences ou tableau des dissimilitudes.
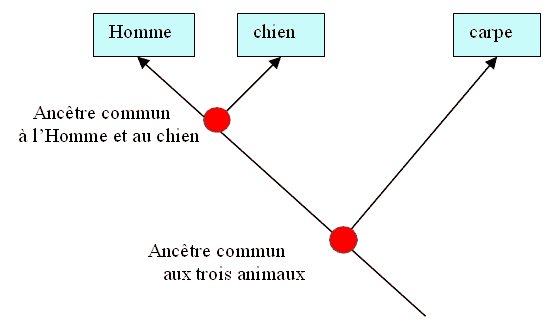
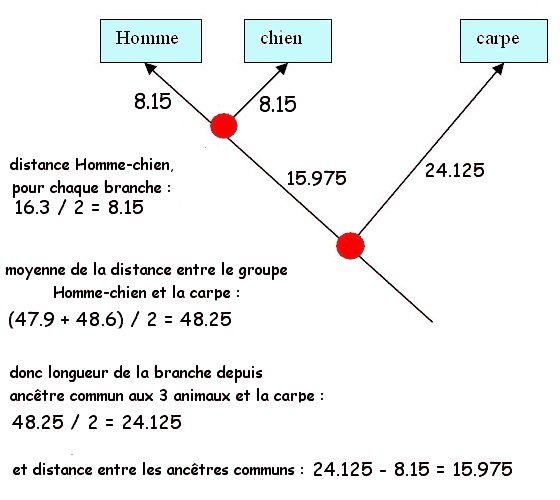
On remarque que la parenté moléculaire est plus grande entre l'Homme et le chien : Ce sont les deux espèces dont les globines a présentent le moins de différences. Les Hommes et les chiens sont plus apparentés entre ex qu’avec la carpe. On en déduit qu’il existe un ancêtre commun à l'Homme et au chien plus récent dans le temps que l'ancêtre commun aux trois animaux. Les lignées Homme-chien se sont séparées plus tard que la lignée carpe avec ancêtre commun à l'Homme et au chien.
Remarques : Si on utilise un vocabulaire rigoureux, il ne s’agit pas d’un arbre " phylogénétique " au sens strict mais d’un arbre " phénétique ". En effet, cet arbre correspond à une comparaison brute des molécules sans se poser la question des possibles convergences moléculaires dues à des mutations reverses : en effet, même si le cas est statistiquement rare, il est possible qu’une mutation intervienne sur un nucléotide déjà changé et permette de retrouver la séquence initiale. Exemple : ATTACGGCT devient AATACGCT par une première mutation, puis redevient ATTACGGCT par une seconde mutation. Il y a un retour à l’état initial ou ancestral de la séquence. De plus, à cause de la redondance du code génétique, certaines mutations de l’ADN ne se répercutent pas dans la séquence du polypeptide traduit (on parle de mutations silencieuses) : dans ce cas, la comparaison de séquences polypeptidiques peut (et c’est fréquent) cacher certaines évolutions. Il est donc préférable de comparer des séquences nucléotidiques plutôt que des séquences polypeptidiques. La comparaison des molécules permet d’accéder à des valeurs chiffrées (ce qui n’est pas le cas des comparaisons au niveau des organes qui sont davantage " qualitatives " avec des résultats en terme de présence et/ou d’absence de tel ou tel caractère homologue). Il est donc possible de tenir compte de ces valeurs pour tracer des arbres dont la longueur des branches (qui représentent les liens de parenté) indiquera une distance génétique : plus la branche est longue, plus la distance est grande.
Des recherches avec des molécules peu évoluées (ADN ribosomal, cytochrome C : pigment respiratoire) on permis de remonter la phylogénie jusqu'aux bactéries. C'est l'association des différentes méthodes qui permet d'établir des phylogénies les plus proches possibles de la réalité possible de l'évolution.
|
||||||||















|
28/04/2008 |
__________ |
|
__________ |
|